| designation: | D1-009 |
|---|---|
| author: | andrew white |
| updated date: | September 26, 2025 |
| human cell atoms: | |
| FLOPs for human cell: | |
| expected simulation year: | 274 |
| power requirement | 200 TW |
abstract: The people want a virtual cell. So let's simulate an entire human cell atom-by-atom for 24 hours with molecular dynamics. I estimate it will take FLOPs and 200 TW of electricity. Based on historic frontier simulations, we should be able to reach this milestone in 2074. I based on this analysis of about 650 historic molecular dynamics simulation papers and some empirical measurements of FLOPs required. See details below.
all atom virtual cell
There has been coalescence around a new term called "virtual cell."1 The virtual cell is a banner definition that captures a federated set of research directions on modeling parts of cells.2 This has led to new funding calls,3 competitions,4 and discussion about what is a virtual cell (and what is it good for).5 The term is a bit vague and has led to some pushback.6 For example, some mean virtual cell to be a "high-fidelity simulation"7 and others are interested in machine learning models for a cell8.
Most interest has been on something with AI for modeling a virtual cell. For example, a recent opinion article from senior authors in the field called for building an AI virtual cell that is "a multi-scale, multi-modal, large neural network–based model that can represent and simulate the behavior of molecules, cells and tissues across diverse states."2 I found it a bit too abstract, but then Abhishaike Mahajan's wrote a nice summary of the various practical ways that such a model can be used.5
An AI virtual cell, as defined above, should require an enormous amount of data. And I'm not sure what it would still be very predictive. Many academic groups and companies have been trying to make predictive models of cell morphology and gene expression under chemical perturbation for years (e.g., Recursion, insitro, Anne Carpenter). They have great models with predictive power, but those we haven't seen any significant effects in drug discovery productivity. It's good science, just not fundamentally changing how biology is done.
a more precise virtual cell
An alluring alternative to an AI virtual cell is to use physics-based simulation to model cells. Namely, molecular dynamics (MD). MD should require less data. It should give us an extremely detailed model of a cell with almost no simplifications. MD has been well-used for modeling proteins and RNA.
Can we simulate an entire cell at atomic resolution?
Anyone who works in the field of molecular dynamics will immediately hate this idea. What are the starting conditions? What is the force field? What about electrons? How will you model the cell membrane? Will it have periodic boundary conditions? How do we assess convergence? Which of the 50 competing water models should we choose?9
Let's ignore all these questions and just try to build a huge fucking simulation and see what happens.
the goal
The goal system sizes are:
- smallest possible cell: atoms for 1 millisecond (JCVI-syn3A) 7
- a typical human cell: atoms for 24 hours
To narrow the scope of this a bit, I will make a few assumptions:
- We're ignoring accuracy details such as force fields, treating reactions/electrons, etc.
- We do not need to consider anything outside of the cell, including solvent
- We have linear scaling in atom count and time
The last assumption is a bit nuanced. Molecular dynamics simulations are fundamentally n-body - meaning you need to consider all n-bodies interacting with all others - . Through many years of algorithmic work, this has been reduced to nearly-linear scaling, mostly via Ewald summations and particle neighbor lists. These work via two principles: interactions between atoms are relatively local and condensed matter (liquids and solids) is mostly at constant density of atoms.
The linear scaling does get more difficult when it comes to long-running simulations. The memory bandwidth becomes too high and so you cannot really get linear scaling once you're breaking up a single group of particles, because you then need to communicate forces. However, the existence of the Anton supercomputer shows that you can redesign chips to make long-running simulations scalable and feasible.10 Thus, I'm going to assume we can get linear scaling in both time and scale of the simulations.
historic trends
Before getting into first principles FLOPs calculations, let's look at the progress on molecular dynamics simulations. I downloaded about 650 research papers on molecular dynamics over the last 35 years and ran them through gemini-2.5-pro to report on simulation duration and number of atoms. I used the following prompt:
- Did the paper do an all-atom explicit solvent molecular dynamics calculation of a biomacromolecule or protein complex?
- If so, how many atoms were simulated
- What was the total simulation time in nanoseconds
The methods may be complex or involve multiple systems. Use your best judgement to choose a representative number of atoms and approximate total of simulation time. You may write out long-form answers to the above, but if you can determine reasonable estimates for parts 2 and 3, create a csv (containing only one row) with the format:
year,atom_count,simulation_duration_ns,paper_title
Output as a CSV enclosed in triple backticks.
If the document is not appropriate for this task or required information is missing, do NOT generate a CSV.
Let's look at simulation size to see when we're on track to simulate a whole cell:
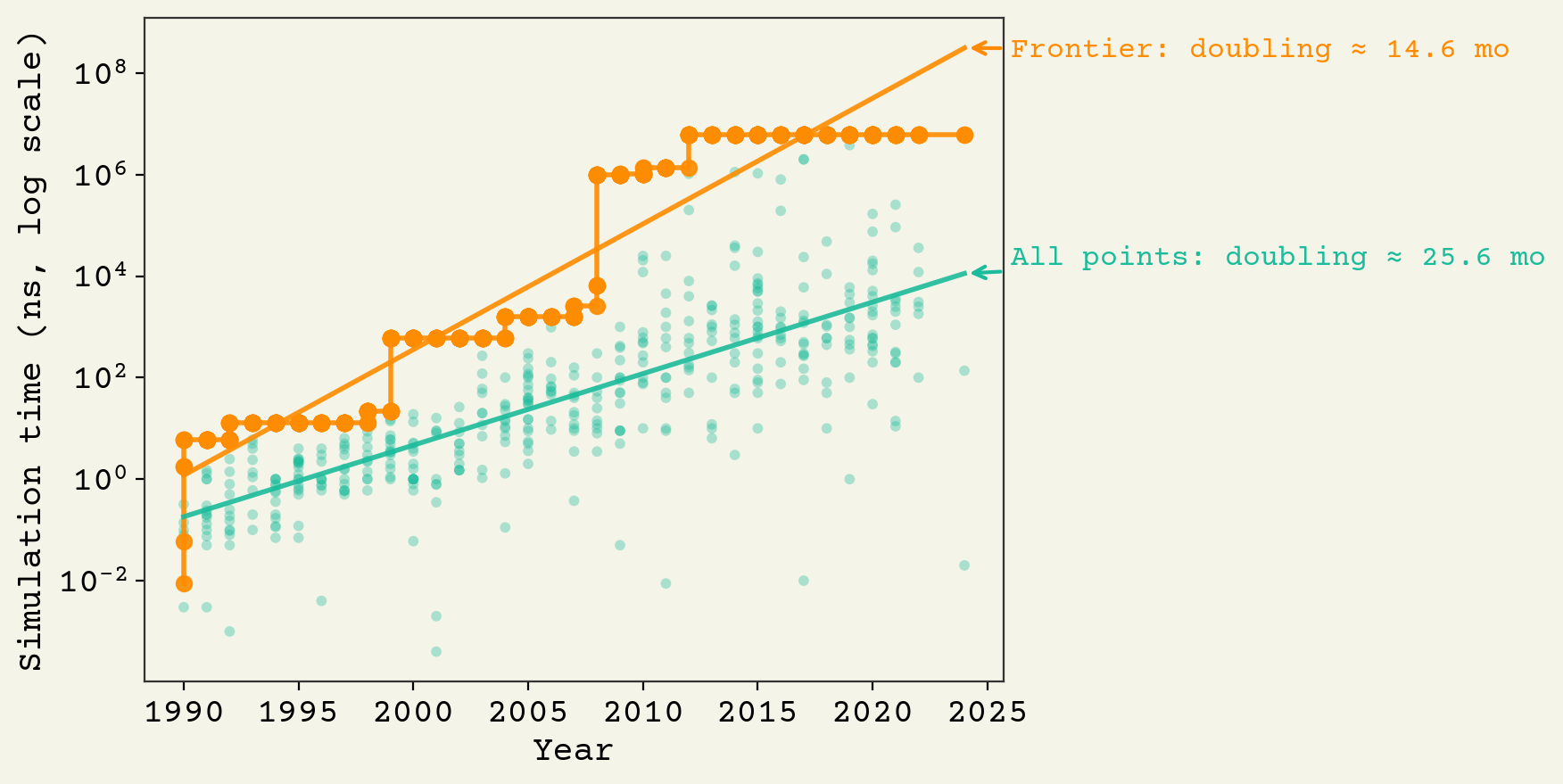
This is on a log y-axis plot, showing the exponential gains in simulation sizes. The doubling-time for the frontier is about 24 months (same as Moore's law). Larger simulations are slightly easier to scale, as discussed above (it is less memory intensive). Thus there is a larger gap in frontier large simulations vs average simulations. With a goal of 10^9 cells in a minimal cell, we've already reached this amount now on the frontier.
There are 5 orders of magnitude left to get to human cells on this chart. So 2057 is when we should expect to see simulations at that size. However, that would probably mean a few nanoseconds of simulation duration. We need to get to 24 hours.
We can apply the same analysis for duration:
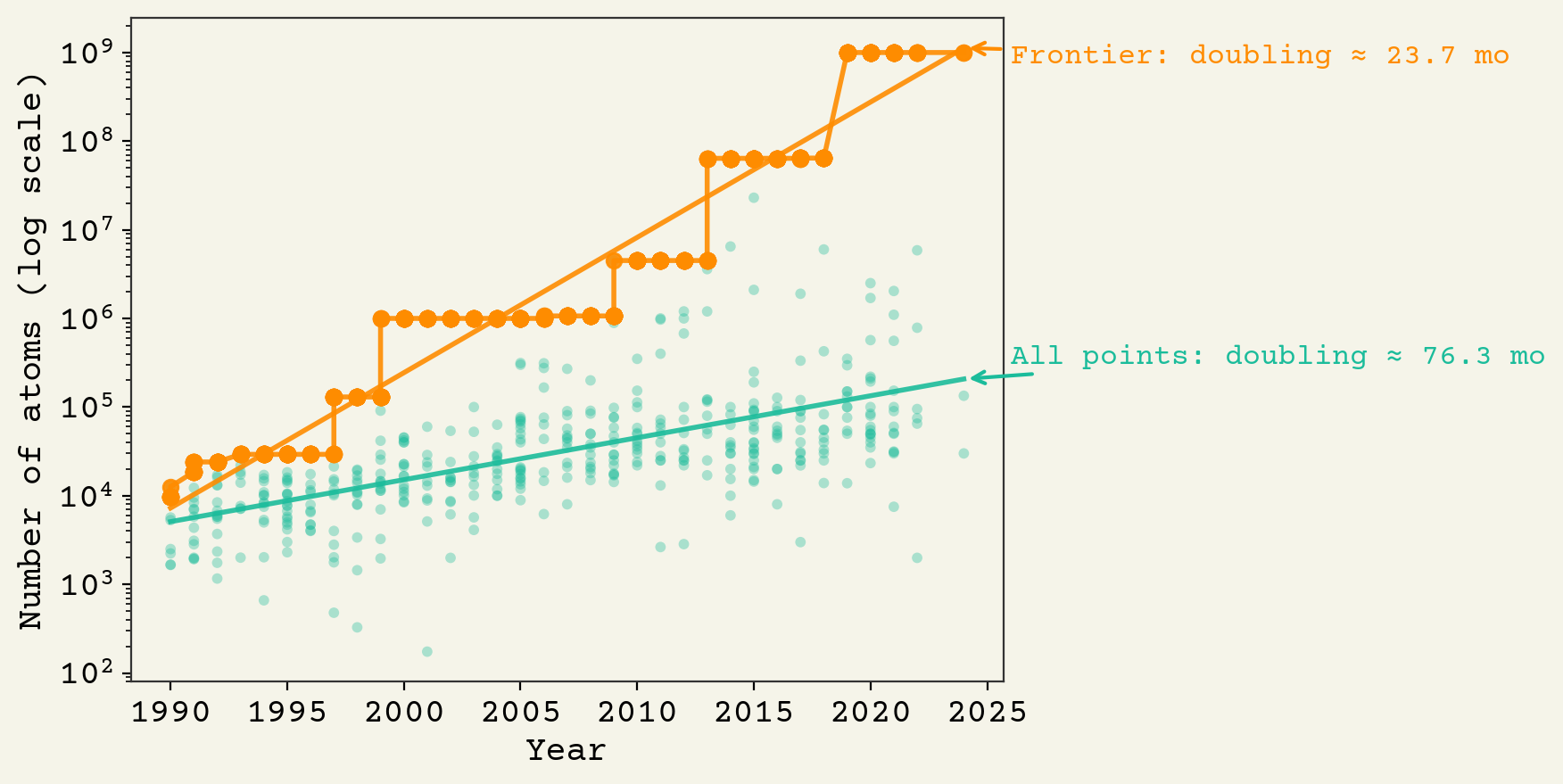
Simulation durations have gone from picoseconds up to milliseconds over last 35 years, with a doubling time of about 14 months. Better than Moore's law is about 24 months (due to algorithms). So there has been progress independent of hardware. These gains come from better algorithms and better utilization of accelerators, like GPUs.
With a goal of 24 hours, we would like to see ns. The highest reported simulations are at milliseconds right now: ns. Based on the trendline, Anton 3 should be reporting a few hundred milliseconds soon (ns).11 We should expect to see simulations at 24 hours in about 25 years: 2050.
For the smaller goal of a minimal JCVI-syn3A cell, we are already at milliseconds.
some math
The above analysis considered simulation size and duration separately, but we need to actually combine them to hit our targets. Let's first find a conversion factor combine the two. We want to convert atomstime into floating point operations (FLOPs).
Let's start by considering the number of interactions for computing the pairwise forces for water at room temperature. Because pairwise forces decay to zero after about 1.2nm, we can consider only the closest neighbors of a given atom. There are about 60 pairwise interactions to consider for water at that cutoff. There are about 25 FLOPs per pair we need to compute the force between them, giving about 1,500 FLOPs per atom per step. This does not include electrostatics, rebuilding neighbor lists, integration, etc. In a typical timing accounting of an MD simulation (relying on personal experience here), the pairwise forces account for about 10--20% of the calculations. So let's just assume about 10,000 () FLOPs per atom per unit time (2 fs).
Let's now try to get some empirical justification for this. I took the timing results from Jones et. al,12 who compared a fixed simulation system across a variety of hardwares for a 23,558 atom system of Dihydrofolate Reductase (which is a common benchmark):
| Accelerator | FP32 FLOPS (TFLOPS) | Engine | Time scale (ns/day) |
|---|---|---|---|
| NVIDIA V100 SXM | Amber | ||
| NVIDIA V100 PCIe | Amber | ||
| NVIDIA TITAN X | OpenMM | ||
| NVIDIA TITAN V | OpenMM | ||
| NVIDIA RTX 3090 | ACEMD |
I have looked up the FLOPS (not to be confused with FLOPs, because FLOPS is per time) per GPU model and put them into the table. If we assume 25% utilization of the GPUs and a 2 femtoseconds timestep, we can convert these to FLOPs per atom per step:
| TFLOPS | time (ns / day) | atoms × time per Day | atoms × time per Second | FLOPs per atom × step |
|---|---|---|---|---|
Our back-of-the-envelope calculation, , matches these empirical numbers across hardware and engines. Very nice to see.
Now that we have a conversion factor of FLOPs per atom per timestep, we can convert our goal numbers to FLOPs. This is an important calculation and I will write it out very explicitly:
In table form:
| System | atoms | duration | FLOPs |
|---|---|---|---|
| JCVI-syn3A | ms | ||
| human cell | hours |
These are pretty large numbers. For example, GPT-4 was estimated to be about FLOPs. is much larger: it would require 100% utilization of the largest ever Project Stargate cluster13 for 150,000 years.
Assuming we don't just start it on our current hardware, we can wait a while for improvements. We can predict when an all-atom simulation of a human cell is feasible based on the historic FLOPs:
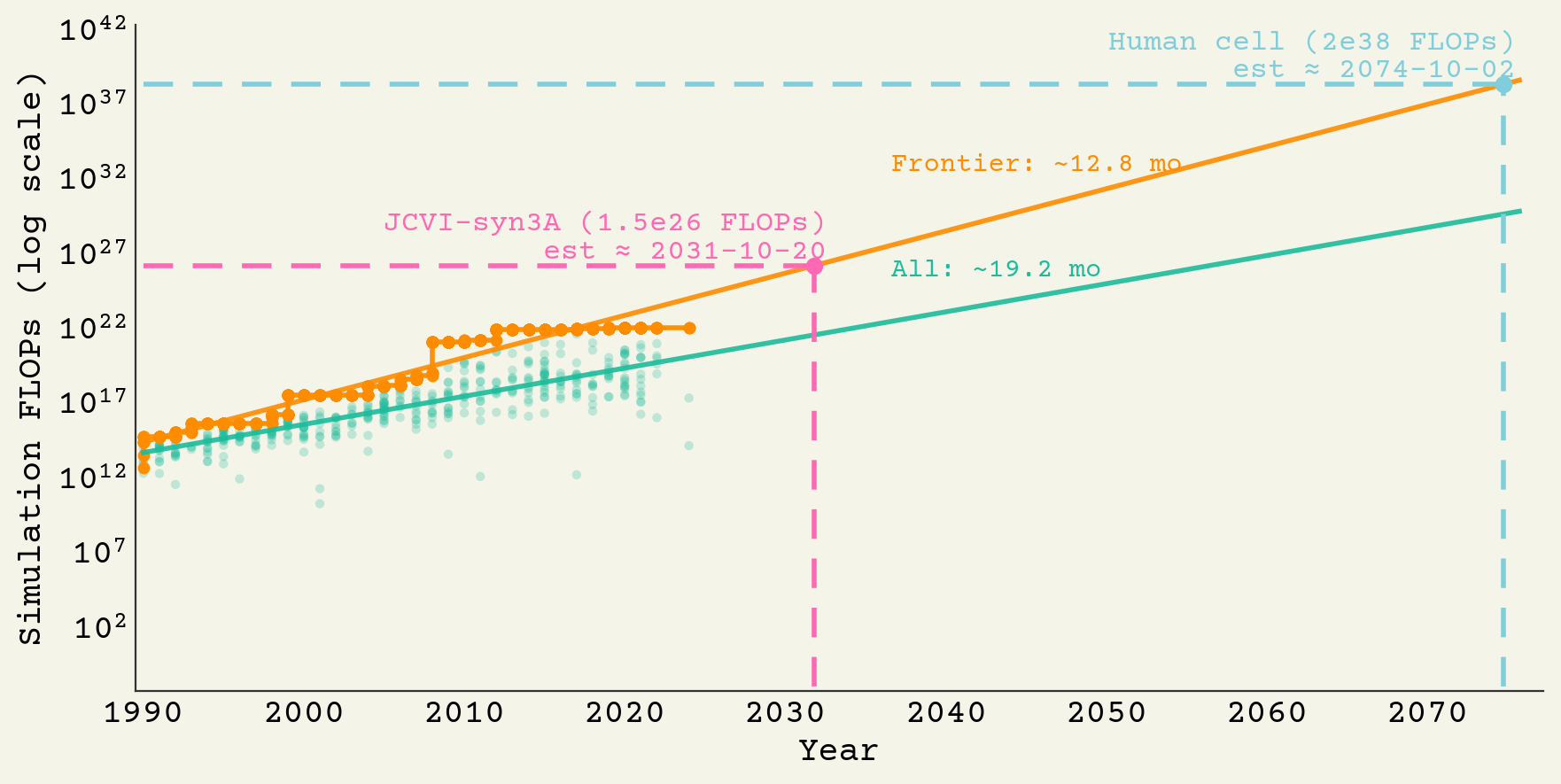
2074 will be the year we can simulate one cell for a day!
engineering the virtual cell
It should be completely feasible to simulate JCVI-syn3A for 1 millisecond today. It's close to the compute required for frontier LLMs, except it's in FP32. That will run about $1B of GPU-hours. If we can cut down to lower precision, maybe we can get to $50M. If we wait for typical frontier simulations to catch-up, so we're not spending $1B, we can get there in 2031.
Unfortunately, we will have to do a bit more engineering work to achieve our goal of human cells. Let's compute the amount of energy it will take to simulate one human cell.
Since we're dealing with 50 year horizons, we need to make some assumptions about what compute looks like in 50 years. The Laundauer Principle limits how much heat we must move around per floating point operation and places limits on possible energy efficiency. If we assume we can achieve 1% of this efficiency limit, we should be able to achieve about in an optimistic scenario.13 This efficiency is about 6 orders of magnitude better than the current most efficient compute, and about 9 orders of magnitude away from current GPUs.
If the simulation is conducted over 6 months of time, the compute energy required is
So we need to supply to the simulation for six months. This is probably feasible - it's about 20x the entire world energy generation in 2025. Hopefully we can devote 20 Earths of electricity to this cause by 2074. Or we can do the simulation over 10 years at the current terrestrial energy production instead of 6 months.
Just growing a human cell, by the way, requires about W.
discussion
Cells are really big and have a lot of atoms. Femtoseconds are really short.
If we were to accomplish this civilization-wide effort, it would still not have any chemical reactions. Chemical reactions are essential for a cell to actually convert molecules, metabolize, and do gene regulation. Almost all methods that model electron transfer require much more compute, and usually do not have linear-scaling in system size. And we skipped over a lot of details about force fields and starting conditions and barostating the membrane. This would be such an insane project.
What would be the benefit? Hard to say. Could be nothing, because it's unknown if molecular dynamics force-fields are equipped to work at this scale. I can think of many more beneficial activities with the compute, so there is definitely an opportunity cost to this project.
conclusion
When you next grow a cell, think about how this would require 20x all of earth's current power to simulate. Biology experiments are very powerful.
An all-atom simulation of human cells is fun to think about. But we probably won't see it until the next century at the earliest.
Footnotes
-
https://www.cell.com/cell/fulltext/S0092-8674(24)01332-1 ↩ ↩2
-
https://chanzuckerberg.com/science/technology/virtual-cells/ ↩
-
https://www.owlposting.com/p/how-do-you-use-a-virtual-cell-to ↩ ↩2
-
https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2023.1106495/full ↩ ↩2
-
See my other blog post about this: https://diffuse.one/p/d1-001 ↩ ↩2